Candidate health checks and risk-benefit assessments
Julian Northen - Technical Director 
Julian Northen - Technical Director
Our previous blog focusing on preformulation studies illustrated how these investigations, along with physicochemical property optimisation, are critical to the successful development of a modern drug candidate. These early investigations provide essential information about how a compound behaves and help identify potential risks before full-scale development begins.
In this blog, we build on that foundation to explore how candidate health checks and risk–benefit assessments use preformulation data, particularly solid form understanding, to guide decision-making as development progresses.
The role of solid form in preformulation
One of the most important aspects of preformulation is understanding the solid form of the compound, which has a significant impact on properties such as melting point, hygroscopicity, solubility and powder flow. Before a form can be selected, candidates should be profiled in terms of predicted solubility, pKa/Log D and permeability. This is often accomplished in silico as part of the lead selection process.
However, this approach often overlooks considerations such as morphology, material handling and behaviour during manufacture. While this is not surprising, it highlights the need to consider such attributes – or desirable critical quality attributes (CQAs) – as development progresses and dose and dosage form are determined.
Establishing a robust preformulation strategy
The basis for robust preformulation requires a strategy and suitable investigation that should consider a number of critical elements (listed in Table 1). This process is iterative, and well-integrated development is preferred, with the understanding that all parts will not be in place until key decisions, such as version selection, have been made.
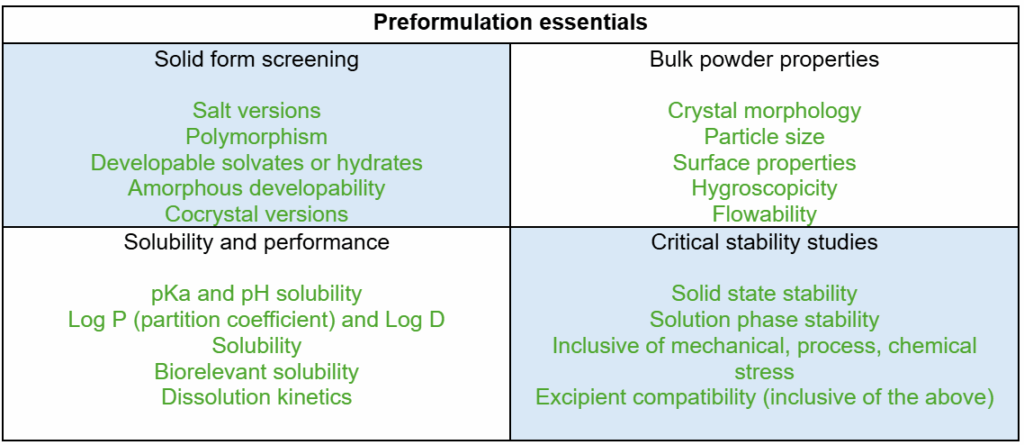
Table 1 – Essential data that is required for effective preformulation risk assessment
All of the elements listed in the table above are, of course, significantly impacted by structure. A large amount of time and effort is invested prior to these studies to determine what a series of viable lead candidates should look like.
From structure to candidate selection
Priority compounds emerge through the systematic elucidation of Structure-Activity Relationships (SARs) through the combined study of potency, specificity and selectivity of analogues based upon a chemical scaffold or common fragment. This work culminates with the identification of a chemical lead, typically where the most potent, specific and selective compound(s) are chosen.
However, relying on this work alone is insufficient, as potency as a path to success is an oversimplification. The critical assessment of the pharmacological properties of the lead molecules, these being Absorption, Distribution, Metabolism, and Excretion (ADME), is equally critical to the selection of an initial lead series and establishes benchmarks against which compounds synthesised during lead optimisation can be evaluated. Concurrent improvements in ADME properties are an integral part of the development pathway, ideally achieved while preserving potency and selectivity where possible.
However, sometimes compounds with lower in vitro potency perform better overall due to improved ADME behaviour. Therefore, the assessment and optimisation of Structure–Pharmacologic/Property–Relationships (SPR) is a further critical step for efficacy evaluation. In addition to assessing compound characteristics such as solubility, protein binding and serum stability, this data allows the development team to prioritise different structural classes and rank-order them not only based on potency but also in relation to potential downstream absorption or metabolism liabilities.
A case study: Potency vs ADME trade-off
As identified by the author during academic study of CDK1/2 analogues, sometimes compounds that are more efficacious overall exhibit lower in vitro potency but better ADME properties. A clear example was the exchange of a chiral alcohol side chain for one bearing a chiral carboxylic acid. The latter was >10-fold more potent (low nanomolar range), but when assessed as the parent compound, it showed a significantly worse ADME profile and was markedly less soluble and less permeable.
Safety and toxicity considerations
One further critical element that directs compound development is safety and toxicity. The majority of early-phase clinical failures and program withdrawals are due to safety concerns or toxic effects. The typical trend as development progresses into later phases is for efficacy to overtake toxicity as a reason for withdrawal. Structure and toxicity evaluation within a lead series is a source of intense study. The causes are not always immediately clear, but may be identified with a robust and integrated team. On-target and off-target toxicity are two important differentiators that impact structure-optimisation choices.
On-target toxicity can be generalised as exaggerated pharmacology induced at the molecular target. Off-target toxicity is characterised by pharmacological engagement at alternative molecular sites. Both are important and require a detailed understanding of moving from in vitro to in vivo models. In particular, understanding species differentiation and predicting the effect in human vs early animal in vivo data can be a significant challenge.
Of the two, off-target activity or promiscuity is a concern that is structurally and physicochemical property-driven and a feature of significant ADME investigation. In fact, promiscuity is readily identified in relevant literature as a route to increased toxicity. Withdrawn and discontinued drugs, when compared with their successful marketed counterparts, more often demonstrate a poor promiscuity profile when reviewed against in vitro pharmacology receptor screens. These are SAR- and SPR- driven safety effects.
Common structural “flags” for toxicity
There are numerous common structural flags from a toxicity perspective:
- Lipophilicity – (High LogP) promiscuity flag, tissue accumulation, cardiovascular and neuro effects
- Basic amine – promiscuity flag, tissue accumulation and at acidic sites
- Reactive structures – genotoxicity – interaction with macromolecules and phototoxicity
However, these features are not automatic exclusions. Many appear in marketed drugs because they are sometimes required for potent binding at the target site of action. Their presence simply warrants closer scrutiny and, where appropriate, mitigation (e.g., reducing exposure, adjusting substitution patterns, or monitoring specific off-targets).
As an example, compounds with a high LogP and low total polar surface area were found to be two times more likely to create a toxic effect (Hughes et al, 2008, Bioroganic Med.Chem.Lett., 18, 4872-5). It is notable that cardiovascular and hepatological effects are two of the major reasons why drugs either fail or are withdrawn from the clinic and market, and for this reason, they form part of the core battery of pharmacological tests listed in ICH S7A.
Linking ADME, potency, and solid form to developability
While ADME, potency and safety are the core drivers for effective lead optimisation and candidate selection, their impact upon the physicochemical properties that manifest as solid form behaviour (and performance) also requires inclusion as part of the overall risk-benefit assessment.
For this reason, there is value in aiding lead candidate differentiation by performing early phase mini screens or ‘health checks’ to verify the developability of a small number of leads. This CMC-biased data does not trump in vivo efficacy or other critical ADMET properties, but is useful where this data is difficult to discriminate between candidates.
In silico prediction vs formal testing
There is no single development route that satisfies every molecule, and studies must be constructed based on data rather than opinion. The pivotal data that forms the basis of such design is robust physicochemical characterisation, which should incorporate, as a minimum:
- Melting point
- Morphology (microscopy)
- Log P
- pKa (including logD for ionisable molecules)
- Chemical stability
- Solubility (pH 1/7.4)
- Permeability
During lead optimisation, a significant portion of these can be estimated using in silico tools like the Modules that are incorporated into software such as SimulationPlus (GastroPlus). An example of these, replicated from the SimulationsPlus website, is illustrated in Figure 1. While this is an extremely effective tool, formal testing is the gold standard for the discrimination and risk-benefit assessment required for accurate selection under the circumstances described.
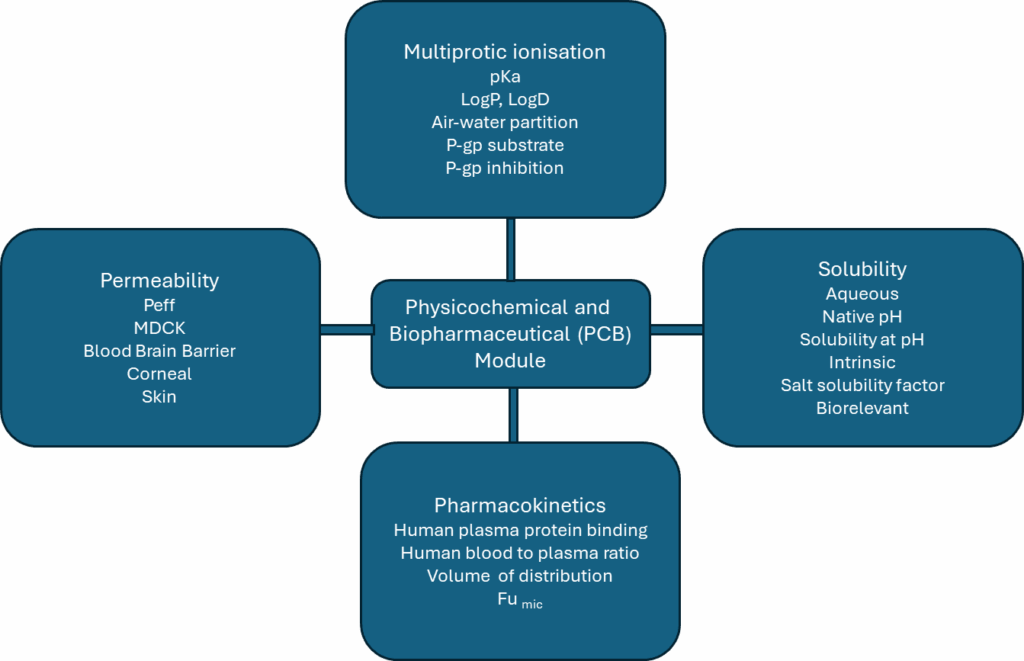
Figure 1 – Data that can be generated in silico using commercial software such as SimulationsPlus
The practicality of mini health checks
Wet testing is often considered costly in terms of material and, during early clinical development, it is often in short supply and high demand for the sort of ADMET testing defined above. However, where the justification exists, mini screens and health checks in fact require very little material to gain a wealth of solid form and performance data relative to the solid form presented.
Individual tests that include partition coefficient, LogP, LogD, pKa and solubility can be performed using milligrams of material. Solid form characterisation can be achieved using milligrams of material, and tests such as XRPD, being non-destructive, can be recycled into thermal analysis and thermal modulation (cycling) experiments to better understand not only the melting point, but thermal transitions. All for the cost of a few milligrams of material, these tests build a foundation that ultimately results in a propensity toward polymorphism statement.
More importantly, if such studies are supported by crystallisation screening and polymorphism screening, aqueous and biorelevant solubility can be benchmarked against a potentially developable crystalline form for each candidate. A depiction of a health check is provided in Figure 2, noting that not all molecules will be treated in exactly the same manner.
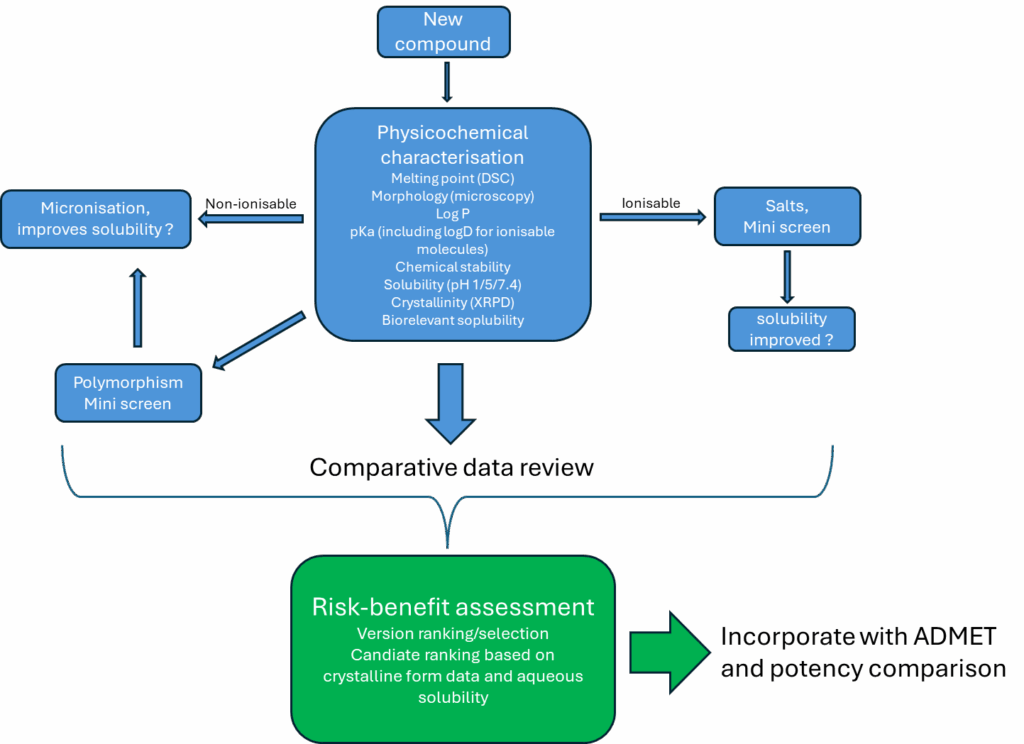
Figure 2 – Simple example of a preclinical preformulation mini screen or ‘health check’ to support early candidate differentiation
Additionally, where candidate molecules are ionisable, a mini salt screen can be incorporated. This will indicate whether the small number of salt formers selected readily form salts, whether the process of formation is challenging, and whether those identified are crystalline or amorphous. It is often the case that such screens are utilised for candidates whose pKa values indicate that the formation of thermodynamically stable salts may be an issue, as it gives a sense of how readily they form and their subsequent characteristics relative to developability.
Once a range of versions or candidate forms is identified, then solubility and biorelevant assessment in parallel is the typical next step to gauge whether solubility and potentially exposure will be improved. Along with ADMET data, this can help define the molecules relative to the Developability Classification System (DCS) and the approaches that may be relevant to achieve a viable formulation or prototype formulations to assess (Figure 3).
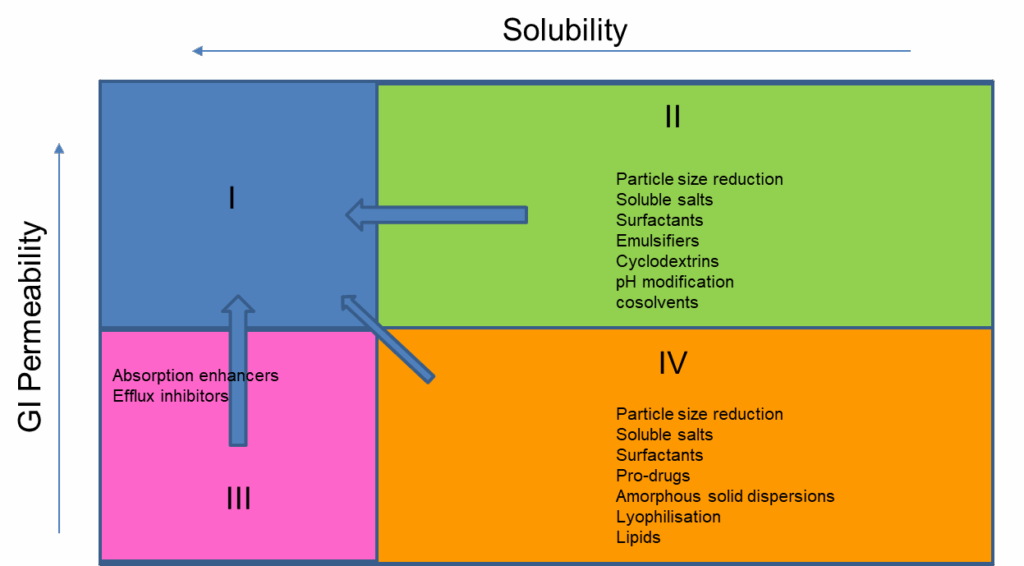
Figure 3 – Example of early pre-formulation considerations to optimise APIs within the separate DCS classes
Integrating data for better decision-making
Whatever the route and testing regime applied, the ultimate goal is differentiation — to aid the selection of a viable molecule at minimal material cost. The purpose of early health checks, preformulation studies, and risk–benefit assessments is to ensure that every decision is data-driven and supported by robust evidence.
As a final note, there is another type of ‘health check’ applied during preclinical and clinical development: in silico solid form validation and assessment. Working from a molecular structure, or ideally, single crystal structural data, a significant amount of supporting information can be obtained.
From such structures, the propensity to form valid salts or cocrystals can be predicted. This information can be used at several points in development:
- At the start of screening, to refine the selection of potential salt or cocrystal formers.
- Mid-development, to extend or adjust a screen where challenges have been identified.
- Later in optimisation, to help differentiate between closely related candidates.
During later-phase studies, structural assessment using molecular descriptors and hydrogen bond propensity tools can also be applied to evaluate whether a more stable polymorphic form is likely to exist. Similarly, in early development, when close analogues show unexpected differences in crystalline form stability, the robustness of the form diagram can be assessed and a risk statement produced for each compound.
Regardless of the size, type, or stage of these supporting health checks, they add significant value when properly designed and applied. When combined with experimental data and early preformulation insight, they enable more confident, evidence-led decisions, ultimately improving the likelihood of successful candidate selection and development.
To learn more about how Onyx Scientific can support your next project, get in touch.