Preparing for process validation and commercial manufacture
Dr Rebecca Matters - Process Development Team Leader 
Dr Rebecca Matters - Process Development Team Leader
As an API advances through the stages of clinical development and overcomes its many challenges, the possibility of commercial production brings both excitement and apprehension.
Even where a process delivers consistent, high-quality material at earlier stages, that consistency alone does not prove it is suitable for commercial supply. To achieve reliable production of high-quality material at a commercial scale, it is necessary to identify the critical quality attributes (CQAs) and critical process parameters (CPPs) that influence them and maintain appropriate control. This process is labelled Process Validation (PV), which is defined by the FDA as the following:
“…the collection and evaluation of data, from the process design stage throughout production, which establishes scientific evidence that a process is capable of consistently delivering quality products”
These activities are split into three distinct stages: Process design, process qualification and continued process verification.
In this technical insight, Dr Rebecca Matters, Process Development Team Leader at Onyx Scientific, explores the early stages of this journey, which lay the groundwork for process validation through the development, optimisation and qualification of a robust manufacturing process. They explore how careful preparation across raw material control, chemistry, analytical development and specification setting supports reliable commercial manufacture.
For this discussion, it is assumed that the synthetic route is fixed and that the process design is both cost-effective and sustainable.
Preparation for process validation
Before formal validation begins, the process design phase focuses on understanding how each element, from raw materials to analytical methods, affects product quality. This foundation ensures that every stage of manufacturing can be controlled, measured and reproduced at scale.
To validate a process successfully, these elements must align both harmoniously and simultaneously. This relies on the collaboration of several areas of expertise, each playing a vital role in embedding quality and control throughout the manufacturing process.
1. Raw materials: Defining the starting point
APIs are often structurally complex. As a result, the overall synthetic route to the final target compound can involve many stages. Impurities and intermediates that appear early in the synthesis are usually removed or transformed during later steps before the API is isolated. For this reason, it is not always necessary for every part of the synthesis to be performed under GMP conditions.
Instead, manufacturers define a synthetic intermediate as the point where formal GMP oversight begins. This compound becomes the regulatory starting material (RSM), the point at which the process must meet the highest quality and documentation standards.
For a synthetic intermediate to qualify as an RSM, it must contribute a significant structural fragment to the API and be fully characterised, meaning its identity, purity and structure are clearly understood. As the RSM directly influences the quality of the API, there must be analytical methods in place to detect any impurities present. The behaviour of these impurities should be tracked through fate and purge studies, which demonstrate whether they are removed, converted or carried forward into later stages. These studies justify the RSM’s purity specifications and ensure the material can be reliably controlled.
However, if any of these impurities affect the impurity profile of the API, the compound is not a suitable starting material due to the impact on product quality. In most cases, commercially available chemicals used in synthesis do not require separate justification as starting materials. However, if one of these chemicals introduces impurities or structural features that influence API quality, it must be treated like an RSM. This means its route of synthesis (RoS) must be defined and its specifications justified based on fate and purge data that show how impurities behave during the process.
2. Chemistry: Optimising reactions and parameters
A reliable manufacturing process depends on the consistency and robustness of each chemical transformation step. Understanding how each reaction behaves under real manufacturing conditions is essential to maintaining quality and reproducibility.
A good starting point for assessing process dependability is a review of previous batch records. These records provide insight into how the process has performed in practice and can help identify any variability, inefficiencies or recurring issues.
Ideally, these batches will be those used for the initial registration filing, as these will have been performed using not only the synthetic route planned for future manufacture but also on a similar scale and at the manufacturing site planned for commercial manufacturing. If issues are found, this evaluation may lead to additional development work, such as refining reaction conditions to reduce impurity formation or adding purification steps to strengthen process control.
At Onyx, process optimisation for commercial manufacturing is carried out using a Design of Experiments (DoE) approach. This structured method systematically evaluates multiple process factors, such as temperature, solvent volume and reagent quantity, to understand their effect on product quality and process performance. By doing so, DoE identifies the CPPs that have the greatest influence on reaction success.
From this analysis, a proven acceptable range (PAR) is established for each key parameter. These ranges define the safe operating window within which the desired CQAs of the product can be consistently achieved. Operating within this defined space allows for minor process adjustments without risking product quality or compliance.
For further details read our blog on how Onyx applies DoE in manufacturing process development.
3. Workup and isolation: Managing process risks
While DoE typically focuses on optimisation of the reaction conditions, the reaction itself is only a small part of the overall manufacturing process. The workup and isolation procedures, where intermediates and the final API are separated, purified and recovered, are also critical to controlling product quality. Therefore, it is important to identify any additional critical parameters in the entire manufacturing process.
To do this, Onyx uses failure modes and effects analysis (FMEA), which examines the impact of different types of failure (e.g., equipment failure, human error) on pre-defined quality characteristics. Every step of the manufacturing process is evaluated to identify where failures could occur and how severe their consequences might be.
Each potential failure mode is scored according to three key criteria:
- Severity: The impact of the failure on product quality/yield or on the overall safety of the process.
- Probability: The likelihood of the failure occurring.
- Detection: How readily the failure is identified.
These scores are then combined to give the overall risk level for this failure mode. An example is illustrated in the table below:
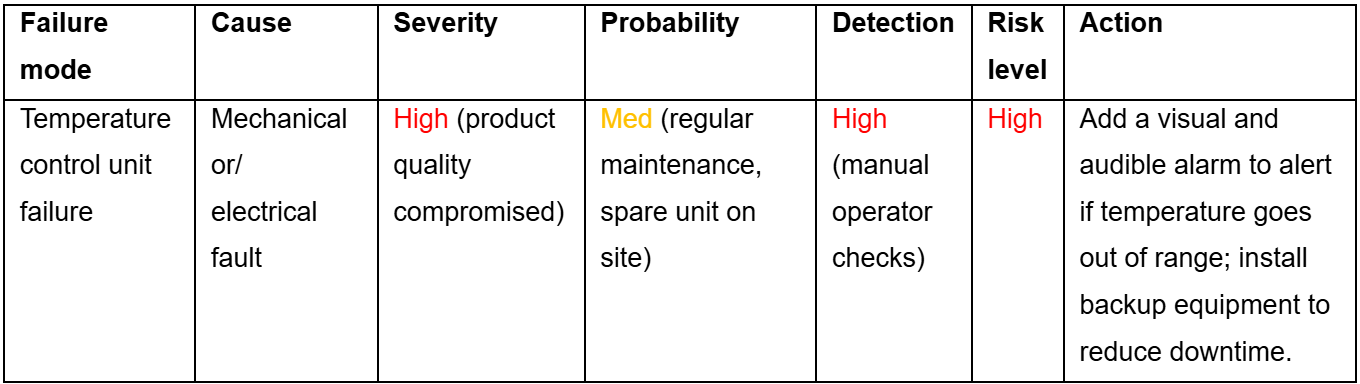
As many of these failure modes may not have been encountered previously in the manufacturing process, accurately assessing the severity may require additional experiments to be performed to confirm the outcome. If the risk level is sufficiently high, additional measures or controls will be put in place in order to reduce the risk to an acceptable level.
By systematically identifying and addressing risks, the FMEA approach enhances control across the manufacturing process. This leads to greater process robustness, reproducibility and confidence that every batch will meet the same high-quality standards.
4. Analytical: Ensuring method suitability and control
In parallel with chemical process development, the analytical methods used to support manufacturing must undergo the same level of careful evaluation. These methods play a central role in ensuring that every stage of production is well understood and controlled.
It is essential that the methods used are able to detect all impurities and degradants present, not just in the process intermediates and API but also in any raw material used in the process. For the API, intermediates and key raw materials, the development of bespoke methods is usually required. In contrast, for more general materials such as solvents or reagents, standard compendial methods (for example, those listed in the USP) may be acceptable. However, these methods must still be assessed to confirm they are suitable for their intended use in the process.
The suitability of an analytical method will depend on the sensitivity required, which may be contingent upon how the raw material is used in the manufacturing process. While a solvent may be commercially available in ≥99% purity by GC, as it is typically used in a large excess compared to the starting material, the remaining <1% of impurities would be present at a much higher level in the overall reaction mixture. Depending on the identity of these impurities, the product quality may be compromised. The type of impurity also matters; for instance, N-methylpyrrolidone (NMP) may contain trace amounts of methylamine, which is a reactive species that can interfere with certain reactions due to its nucleophilic and basic nature.
To ensure reliability, all analytical methods used in the process must be validated according to ICH Q2 guidelines. Validation confirms that each method is accurate, precise, specific and suitable for its intended purpose.
For a more detailed discussion of analytical method validation, see https://onyxipca.com/news/a-practical-guide-to-forced-degradation-and-stability-studies-for-drug-substances/.
5. Specifications: Establishing performance limits
Once the manufacturing process is well understood and the analytical methods have been proven suitable, specifications for process intermediates, in-process controls (IPCs) and the final API can be clearly established. These specifications define the acceptable limits for purity, potency and other critical quality attributes, ensuring that every batch meets consistent standards.
The specification limits should be based on historical batch data, which provides evidence of typical process performance, and further justified through fate and purge studies that demonstrate how impurities are formed, transformed or removed throughout the process. This combination of practical experience and scientific evidence ensures that each specification is both realistic and defensible to regulators.
To provide further confidence that these specifications are appropriate and that every aspect of the manufacturing process performs as expected, it is prudent to perform the optimised process on the scale and using the equipment planned for future manufacture. The success of this commissioning, or engineering, batch should help assuage any lingering doubts about the process. However, if any issues do arise, it also crucially allows for minor modifications to be made before the manufacturing process and quality criteria are finalised.
Process validation
With the final manufacturing process in hand, accompanied by effective analytical testing and well-defined specifications, the process is now ready for the final phase of evaluation: validation.
For validation, a protocol is prepared that specifies how each step of the process will be performed and the key criteria to be measured. Although committing to such a rigid set of parameters and outcomes may seem daunting, the success of the commissioning batch and the underlying attention to detail during process development and optimisation help set a solid foundation for success.
Each batch of this campaign will be rigorously reviewed both during production by Quality Assurance and, upon completion, the relevant authority as part of a regulatory submission. The focus of both parties is to confirm that all batches followed the stipulated process and fulfilled all of the pre-defined quality requirements. This confirms that the process is under control and capable of reproducible commercial manufacturing. Once validation is complete, continued process verification (CPV) is typically performed with trend analysis used to identify problems early and ensure the process stays under control.
Preparing for success in commercial manufacture
Process validation marks the point where science, control, and experience converge. While preparing a manufacturing process for commercial production can be intimidating, a systematic and diligent approach across multiple disciplines can provide the necessary support for an API in its final steps towards its ultimate therapeutic goal.
Onyx supports clients through every step of this journey, helping transform promising molecules into consistently manufactured commercial products.
To learn more about how our team can assist in process design, optimisation and validation, contact us today.